| |
We develop and apply chemical methods to label proteins and study their function in living mammalian cells. With chemical labeling, a protein of interest is expressed as a fusion to a polypeptide tag that interacts specifically with a synthetic fluorophore or other chemical probe (Figure 1). Synthetic fluorophores can offer distinct advantages over commonly used fluorescent protein labels, such as superior brightness, longer fluorescence lifetime, higher photostability or
red-shifted emission. Chemical probes can also imbue proteins with properties that are not easily encoded genetically, for example environmental sensitivity, photo-crosslinking ability or high electron density. Our experimental approach incorporates synthetic organic chemistry, molecular and cell biology, and microscopy.
Our current research interests include:
1) Multiplexed chemical labeling of proteins in living cells: Molecules that bind specifically and with high affinity to proteins can be
developed into powerful tools for chemical biology. The interaction between substituted 5-benzyl pyrimidines and different forms of dihydrofolate reductase (DHFR) can be exploited for chemically labeling fusion proteins in mammalian cells. For example, the specific, high affinity (KD ~ 1 nM) interaction between trimethoprim and E. coli DHFR has been exploited to develop the LigandLink(tm) Universal Labeling technology (Active Motif, Inc., Carlsbad, CA, www.activemotif.com) that makes it possible to tag eDHFR
fusion proteins in wild-type mammalian cells with cell-permeable trimethoprim-fluorophore conjugates (Figure 1). We are currently developing additional antifolate/DHFR pairs for in vivo protein labeling, with the ultimate goal being the simultaneously labeling of multiple proteins in a single cell.
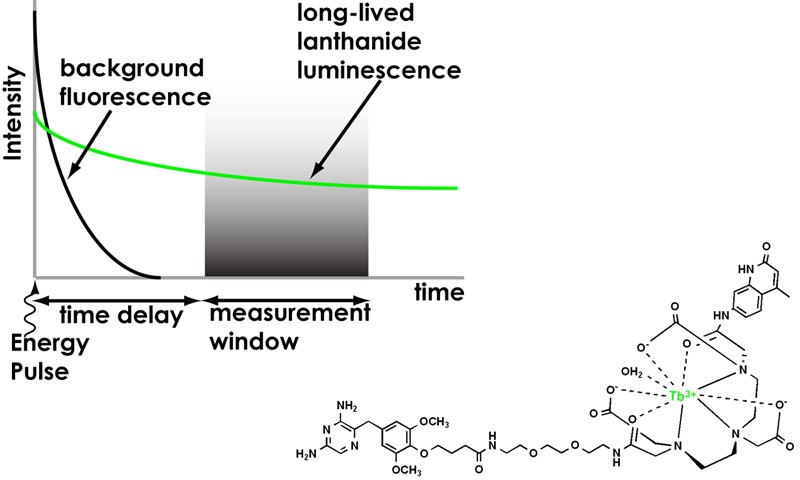
2) Time-resolved microscopy of lanthanide probes. Lanthanide luminescence offers several advantages for fluorescence-based biological assays: 1) large Stoke’s shifts (>150 nm) and multiple, narrow emission
bands (<10 nm at half-maximum) allow efficient spectral separation of emission signals; 2) long luminescence lifetimes (micro- to millisecond) enable time-resolved detection methods to remove scattering and autofluorescence background; and 3) relative insensitivity to photobleaching allows for prolonged detection. We are exploring time-resolve microscopy (Figure 2) as a means of imaging lanthanide probe-labeled proteins at the single-molecule limit of detection.
3) Dynamic visualization of protein-protein
interactions: Transient protein-protein interactions control cell growth and function. We are developing lanthanide-based, protein labels with long (msec) luminescent lifetimes (Figure 2). These probes will facilitate in vivo, time-resolved imaging of resonance energy transfer between interacting proteins. By analyzing the luminescent decay curves of long-lifetime donors, it will be possible to determine the stoichiometry of the interaction, as well as the dynamic localization within the cell. Furthermore, it
will be possible to time-gate out any background fluorescence from endogenous biomolecules, allowing luminescent live cell imaging with unprecedented signal-to-noise ratio.
|
| |
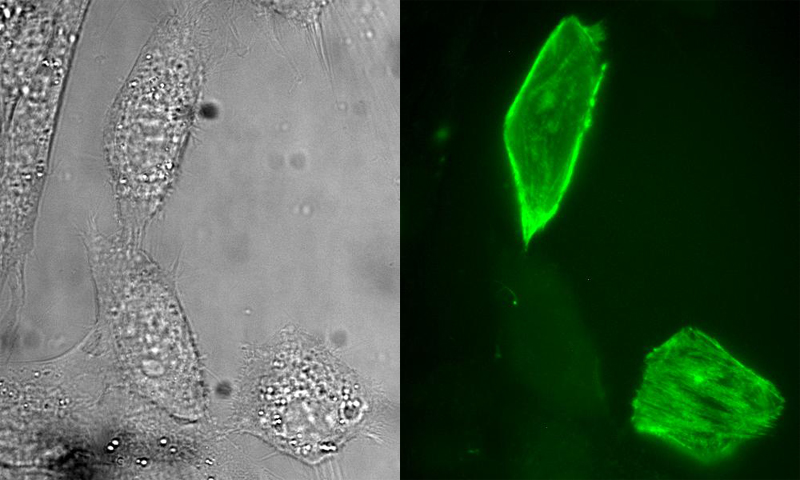 |
Figure 1. Selective labeling of a fusion protein with a small molecule in living cells. Epi-fluorescence micrographs (left, bright field; right, fluorescence) of NIH3T3 cells that were transiently transfected with DNA encoding a fusion of myosin light chain (MLC) to E. coli dihydrofolate reductase (eDHFR). The cells were incubated in growth medium containing a heterodimeric conjugate of trimethoprim (TMP) covalently linked to a fluorescein
|