|
|
Research Overview
Olefin metathesis has become one of the most powerful synthetic arsenals for carbon-carbon formation and the impact of which has been recognized by the 2005 Novel prize in chemistry “For the development of the metathesis method in organic synthesis”. Among metal-carbene complexes that can catalyze the metathesis reactions, ruthenium-alkylidene carbenes known as “Grubbs catalyst” have shown wide range of application in organic synthesis due to their relatively high stability to air and moisture as well as polar functional groups. Most pronounced applications of metathesis chemistry range from synthesis of natural products, pharmaceuticals, and polymers of novel properties. Additionally, the greener nature of metathesis compared to other synthetic methods has additional benefits especially considering the environmental issues. While many different types of metathesis involving alkenes such as ring-closing metathesis (RCM), cross metathesis (CM), ring-opening metathesis (ROM), ring-rearrangement metathesis (RRM), ring-opening metathesis polymerization (ROMP), and non-metathetic transformations catalyzed by ruthenium carbenes (G-I, G-II, GH-II) have been studied extensively, the corresponding metathesis involving both alkenes and alkynes known as enyne metathesis have been underdeveloped. However, significant advances in RCM and its extension to metallotropic [1,3] shift bodes well for the further development of enyne metathesis. [Top]
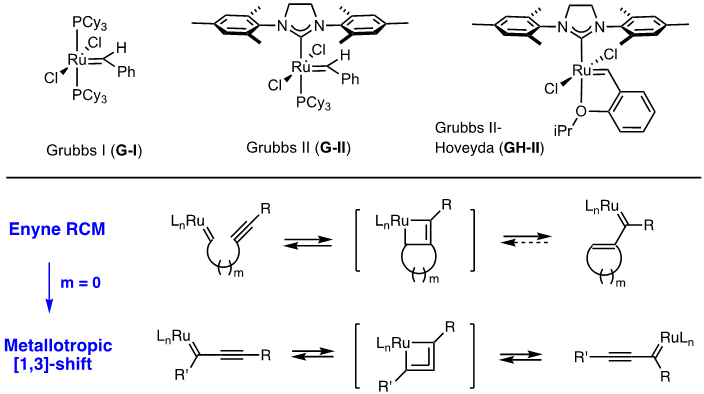
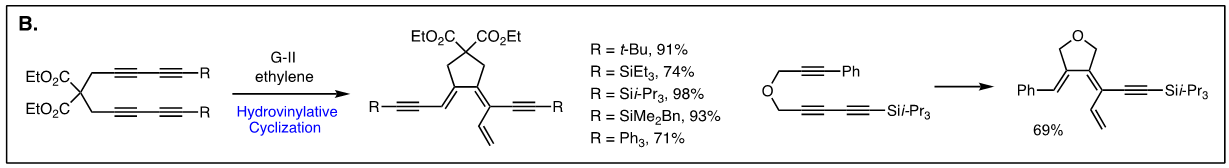
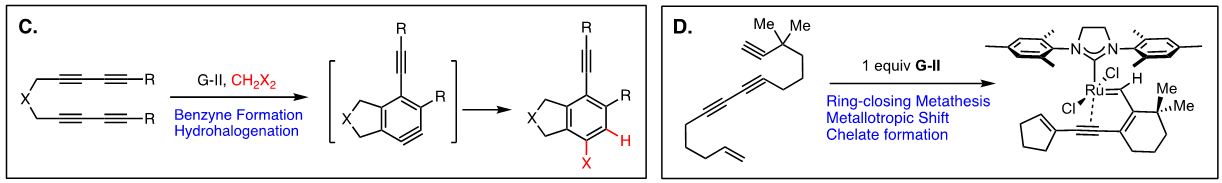
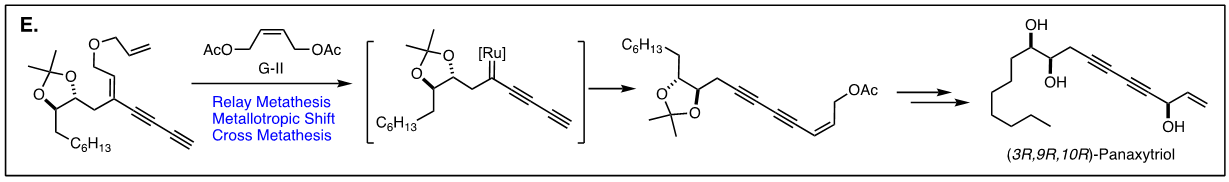
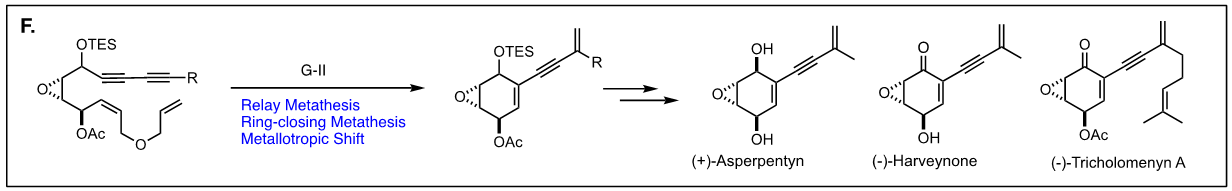
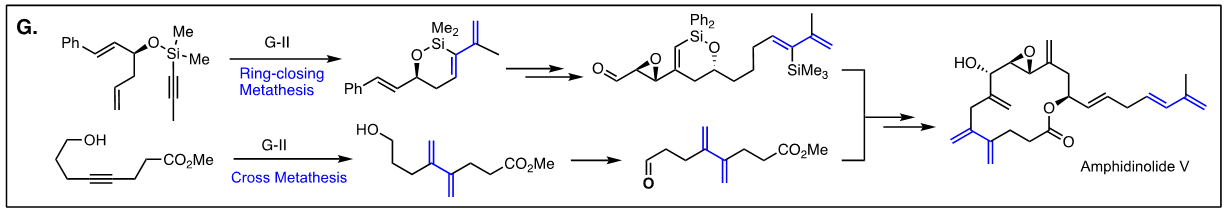
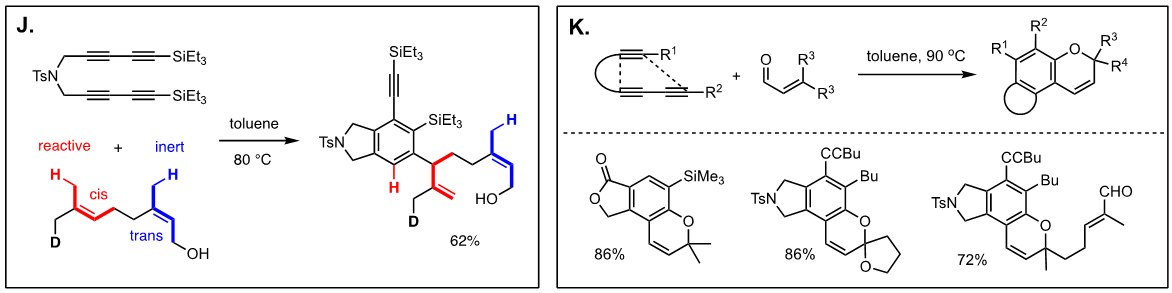
Arguably, the first enyne RCM reaction was reported by Katz and coworkers in 1985 using tungsten Fisher carbene complexes. Since then a variety of low-valent transition metals have been used to catalyze this reaction. However, it was not until the development of ruthenium carbene in the 1990’s that the enyne metathesis is considered to be a viable metathesis tool, yet it still has a narrow substrate scope due to its mechanistic complexity compared to alkene metathesis. In light of this, we have been exploring the tandem bond-forming capacity of enyne metathesis. These endeavors have led to significant advances in our understanding of the reactivity and selectivity of various substrate platforms in RCM and CM reactions. Furthermore, the highly effective metallotropic [1,3] shift of propargylic ruthenium carbene allows for a series of sequential enyne metathesis, which can form molecules of extended conjugation with both double and triples bonds. Tandem metathesis of multiynes with a sequence of enyne metathesis and metallotropic [1,3] shift has led to the discovery of many unprecedented transformations and hidden catalytic activities of Ru-alkylidene carbenes. These novel transformations include the formation of highly conjugated oligo-enynes (A), 1,4-hydrovinylative cyclization of triynes and tetraynes (B), benzyne formation via the hexedehydro Diels-Alder reaction of multiynes followed by transfer of hydrohalogen from common halogenated organic solvents (C), and metallotropic shift-driven formation of ruthenium-alkyne chelate (D). The prowess of the tandem bond-forming nature of enyne metathesis in tandem with metallotropic 1,3-shift has been applied to the synthesis of 1,3-diyne- and 1,3-enyne-containng natural products (E, F). Also, relying on enyne RCM and CM as the key step for the construction of 1,3-diynes, an efficient total synthesis of amphidinolide V was achieved (G). Our current research focuses on the development of new ruthenium alkylidene complexes to further expand and discover novel reactivities of these complexes and their application to the synthesis of natural products and conjugated oligomers. [Top] 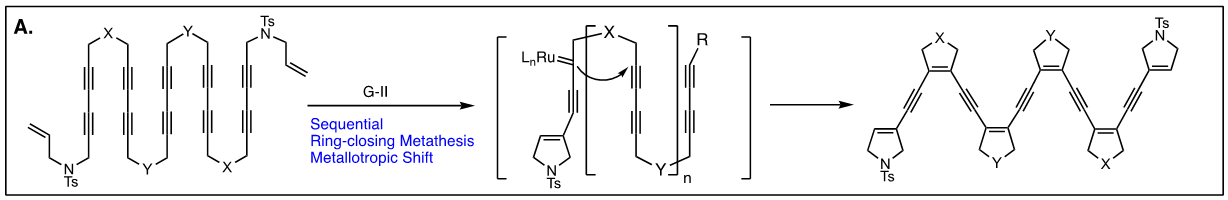
Representative Publications
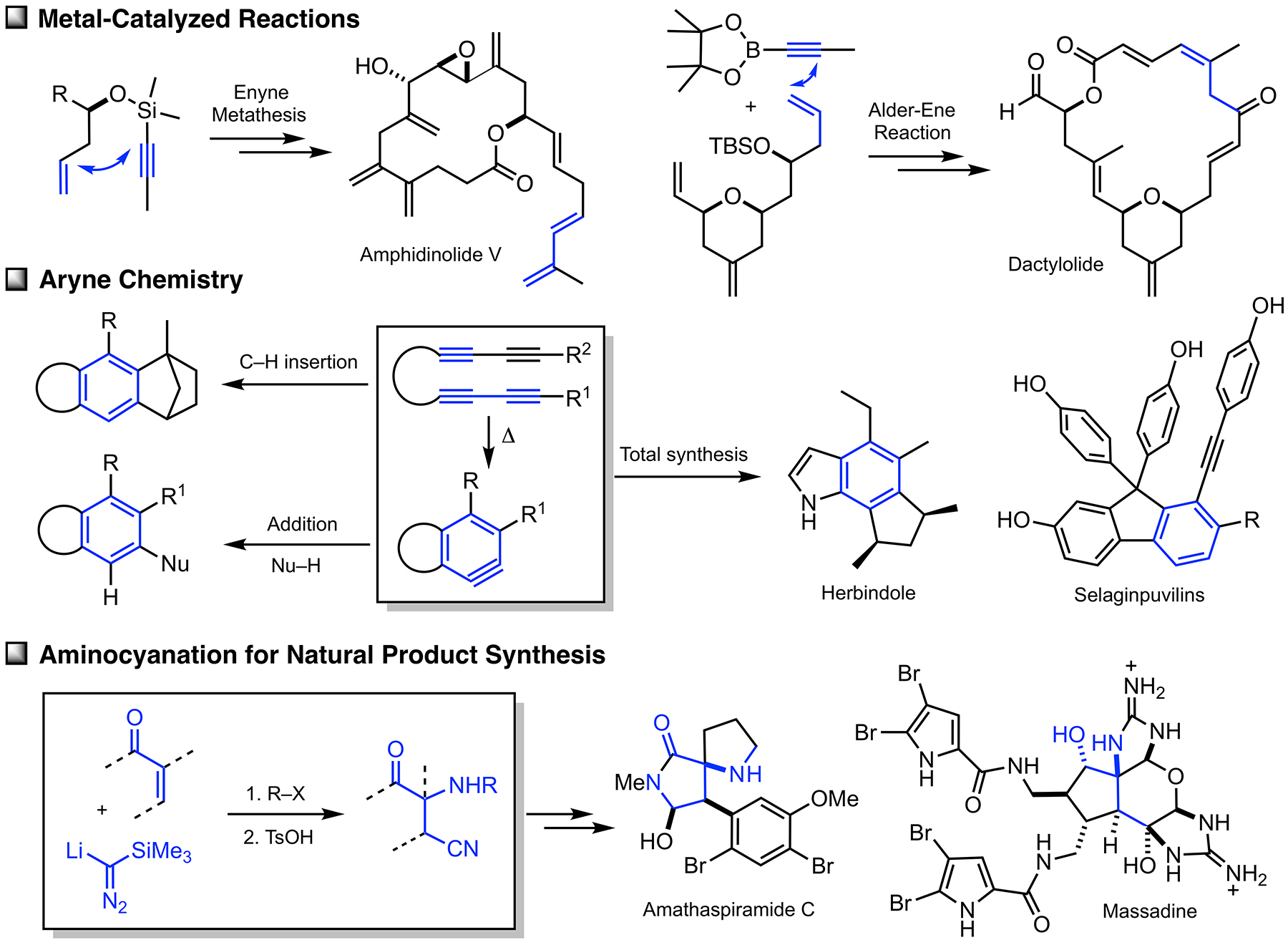
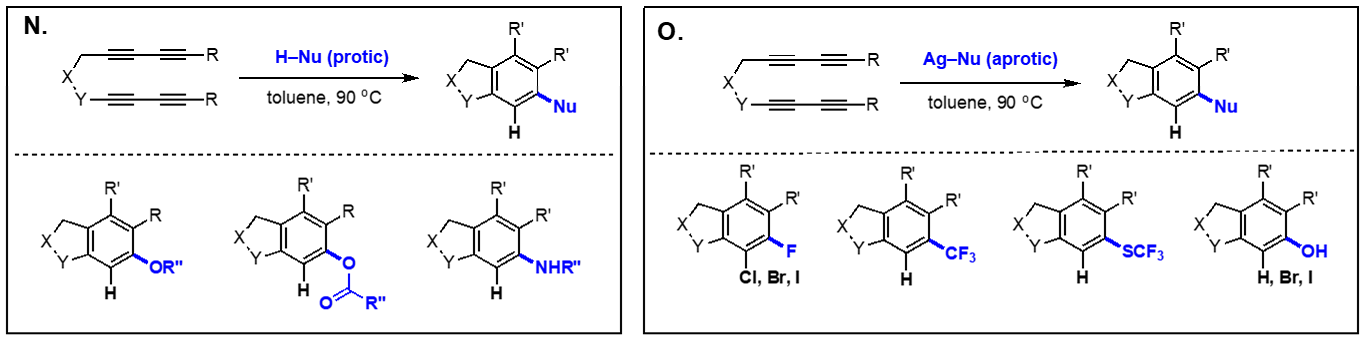
On the basis of the facile formation of halogen-containing arenes directly acyclic tetraynes, we have started to explore the reactivity of arynes generated via the hexadehydro Diels-Alder reaction toward various nucleophiles with both intra- and intermolecular reaction manifolds. Our investigation on the aryne reactivity includes reactions with metal-catalyzed addition of poor nucleophiles and those with relatively good nucleophiles without a catalyst. Representative examples of these transformations we have explored are briefly summarized below. [Top] Metal-catalyzed reactions of arynes: The transformations of arynes catalyzed by a transition metal catalyst include; (A) C–H insertion, (B) inter and intramolecular hydroarylation, (C) intramolecular hydride transfer from a tethered trialkylsilyl group, (D) hydrohalogenation reactions using halogenated hydrocarbons as the source of HX, (E) hydrofluorination using pyridine-HF complex as the source of hydrogen fluoride, (F) mono-addition of nitrile followed by trapping of water to form amides and/or double addition of nitriles to form quinazolines, (G) double addition of isonitriles to generate benzocyclobutene-1,2-diimines, and (H) mixed addition of nitrile and isonitrile to generate 3H-indol-3-imines and 3-iminoisoindolin-1-ols. [Top] 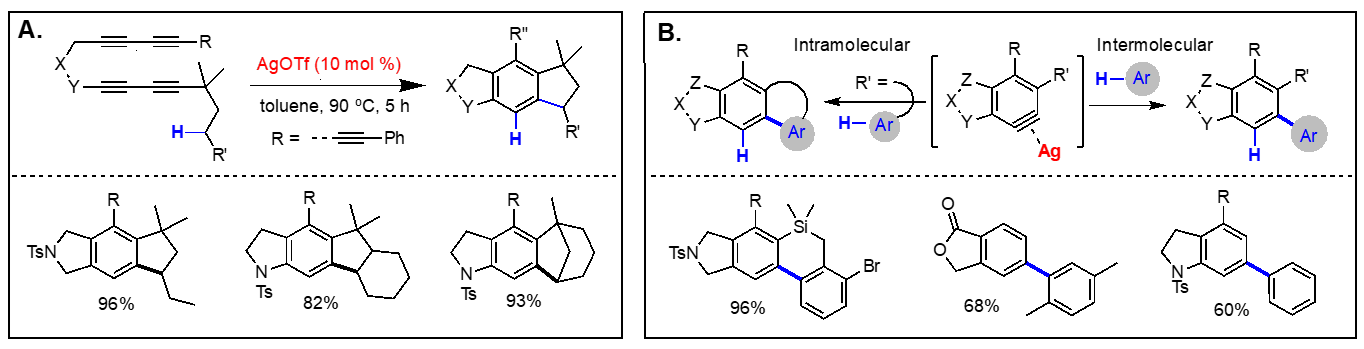
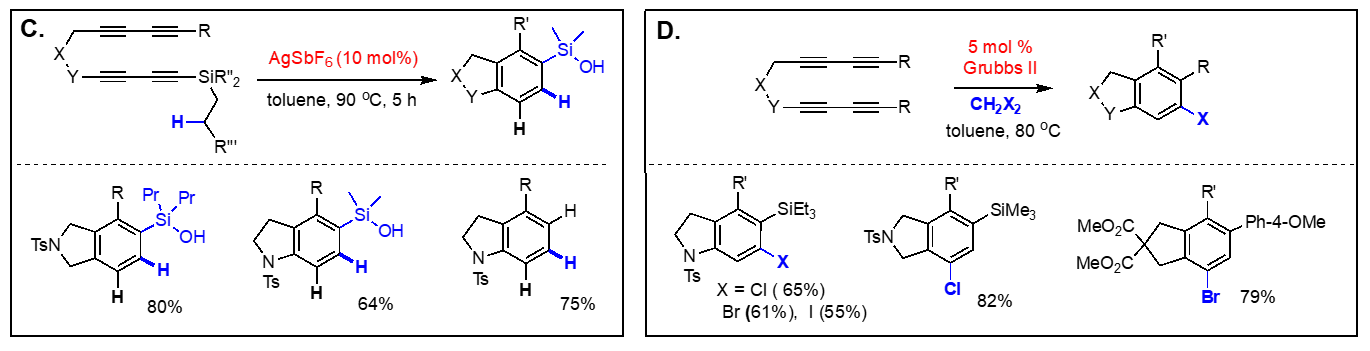
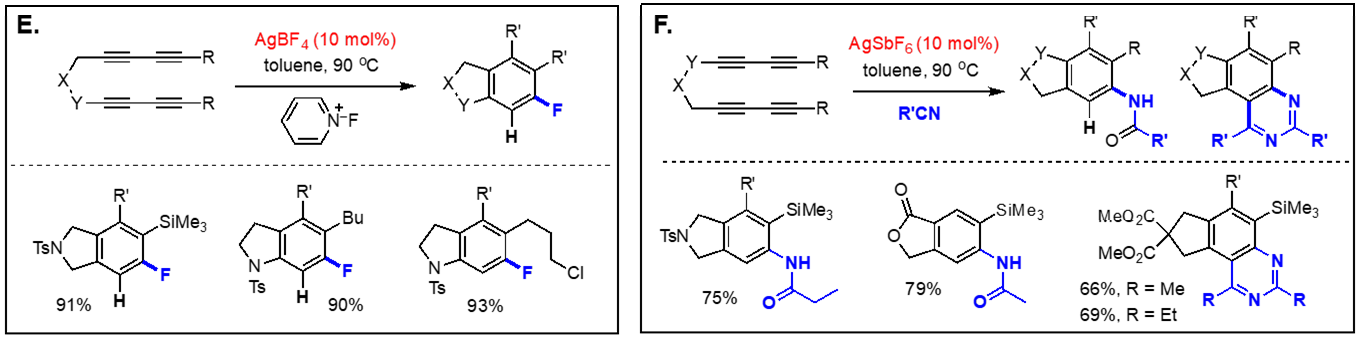
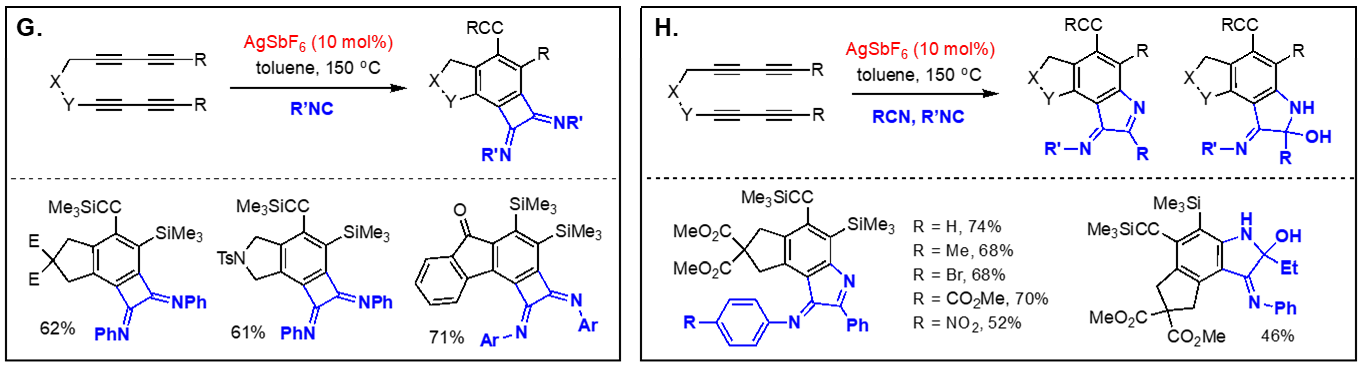
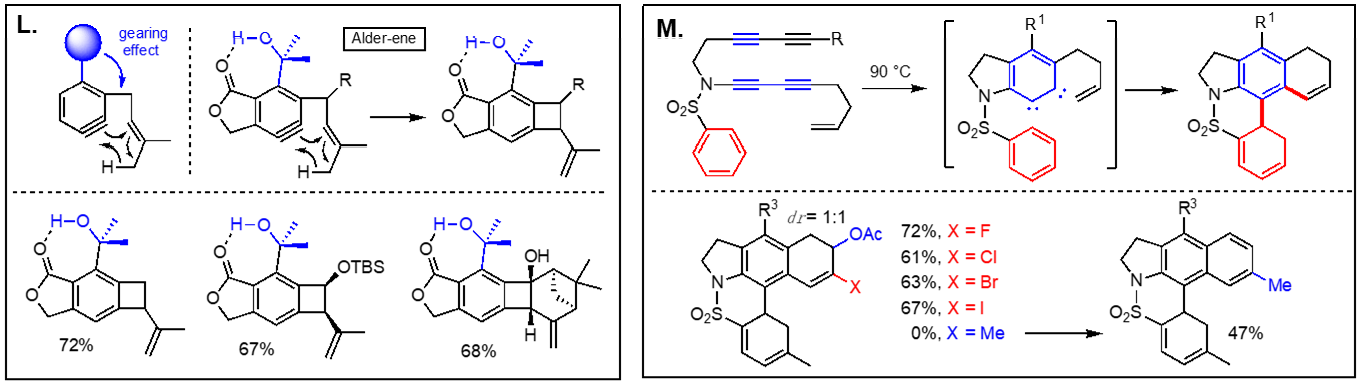
Thermal reactions of arynes: Due to the bond distortion of the alkyne moiety in aryne species, they readily participate in a variety of reactions, including Diels-Alder reaction, Alder-ene reactions, 1,3-dipolar cycloadditions, and nucleophile addition reactions. The representative reactions of arynes generated via the hexadehydro Diels-Alder reactions without using any catalyst involve; (I) Type-I and Type-II Alder-ene reactions, (J) intermolecular Alder-ene reaction with functionalized alkenes, (K) addition of α,β-unsaturated aldehydes, (L) steric pressure-driven ene-reaction to form benzocyclobetenes, (M) aryne-mediated deraromatization, (N) regioselective addition of weak nucleophiles such as alcohols, carboxylic acids, and amines, and (O) addition of nucleophiles containing a silver counter cation including AgBF4, AgCF3, AgSCF3, and AgO2CCF3 to afford the corresponding addition products and phenols. We are expanding these aryne-based addition reactions with other nucleophiles. [Top] 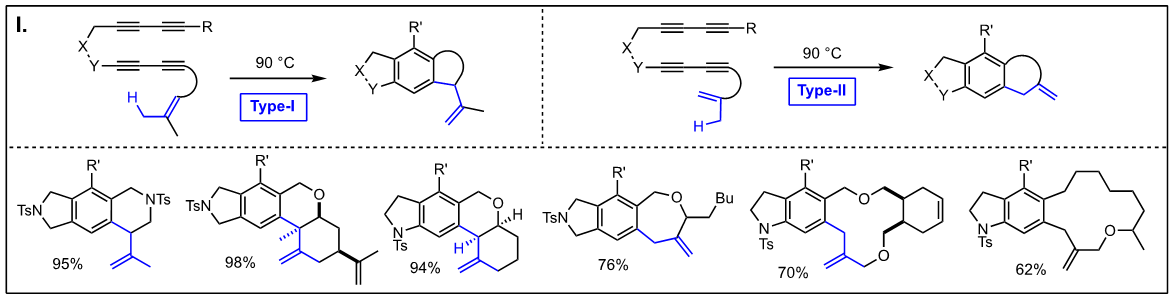
Application of aryne hydrohalogenation to the synthesis of herbindole B: The prowess of aryne-based construction of functionalized arenes was demonstrated by the synthesis of an antifeedant and cytotoxic natural product herbindole B. The key strategy for the construction of the core structure of herbindole B is the mode-selective HDDAR followed by hydrobromination of an aryne intermediate formed from bis-1,3-diyne A1. Bromoarene product A2 was converted to A3 via a three-step sequence involving removal of the tertiary alcohol moiety, hydrogenation, and desilylation of the benzylic trimethylsilyl group. The Sonogashira coupling between A3 and 3-butyn-2-ol followed by acetylation furnished A4. Gold-catalyzed rearrangement of the acetate moiety followed by the Nazarov cyclization afforded A5. The required methyl group was installed via the addition of MeMgI followed by acid-catalyzed dehydration generated indene derivative, which was hydrogenated using the Crabtree catalyst to provide known compound A6. Sato achieved a total synthesis of herbindole B from A6 in two steps, thus a formal synthesis of herbindole B was achieved. Relying on the effectiveness of forming functionalized indolines by arynes, we are pursuing the total synthesis of other indole-based natural products. [Top] 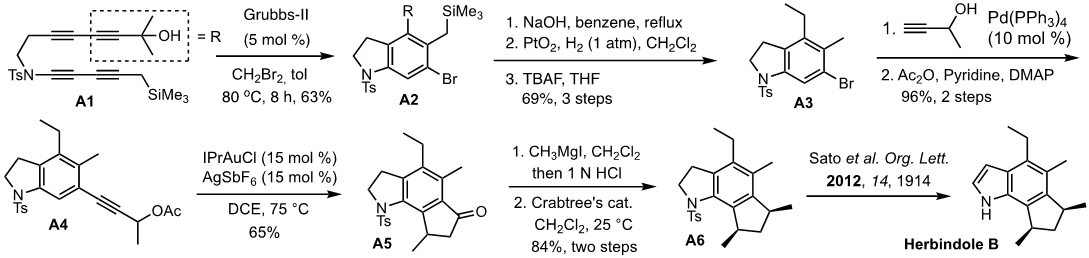
Aryne-based approach to the synthesis of selaginpulvilins: In our effort to illustrate the effectiveness and utility of aryne-based arene synthesis, selaginpulvilins were selected as a synthetic target. From a biological activity standpoint, selaginpulvilins are highly valuable targets because they show remarkable inhibitory activities against PDE4, thus these natural products can be used as a platform for developing selective PDE4 inhibitors. The synthesis of selaginpulvillins is commenced with a Sonogashira coupling of commercially available aldehyde B1 with ethynyltrimethylsilane to generate B2. Merging B2 and B3 followed by desilylation provided triyne B4. A Cadiot-Chodkiewicz reaction with a proper terminal alkyne afforded tetrayne B5. Oxidation of the secondary alcohol in B5 with MnO2 induced a spontaneous formation of an aryne intermediate, which was hydrogenated by cyclooctane, delivering the core of selaginpulvillins B6. Because direct Friedel-Crafts arylation of fluorenon with phenol failed with B6, 4-methoxyphenylmagnesiumbromide was added to the bis-benzylic ketone to generate the corresponding tris-benzylic alcohol B7. Lewis acid-mediated arylation of the tertiary alcohol was found to be problematic with B7-a containing an acetoxy group at the benzylic position, which yielded bis-phenol adduct B8 in low yield. On the other hand, desired Friedel-Crafts arylation occurred with B7-b containing a trimethylsilylmethyl substituent, which delivered B9 in good yield. Removal of the trimethylsilyl group afforded O-trimethyl-protected form of selaginpulvilin C, and further treatment of this compound with MeMgI (neat) at elevated temperature (160 °C) finally afforded selaginpulvilin C. The first total synthesis of this natural product (9-step, 22% overall yield) clearly illustrates the prowess of HDDAR as the tool to construct highly elaborated fluorene framework from of a relatively simple acyclic multiynes. We are currently pursuing the synthesis of other selaginpulvilins from B9. [Top] 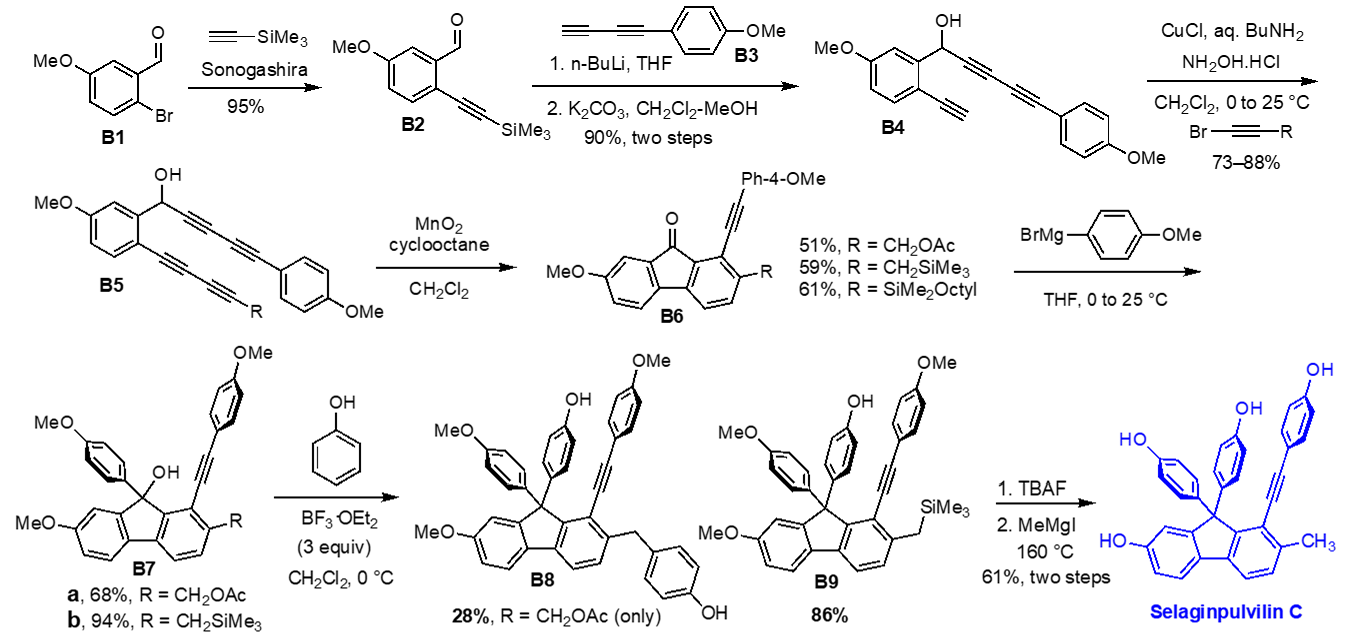
Representative Publications
New Reaction Development with Trimethylsilyldiazomethane
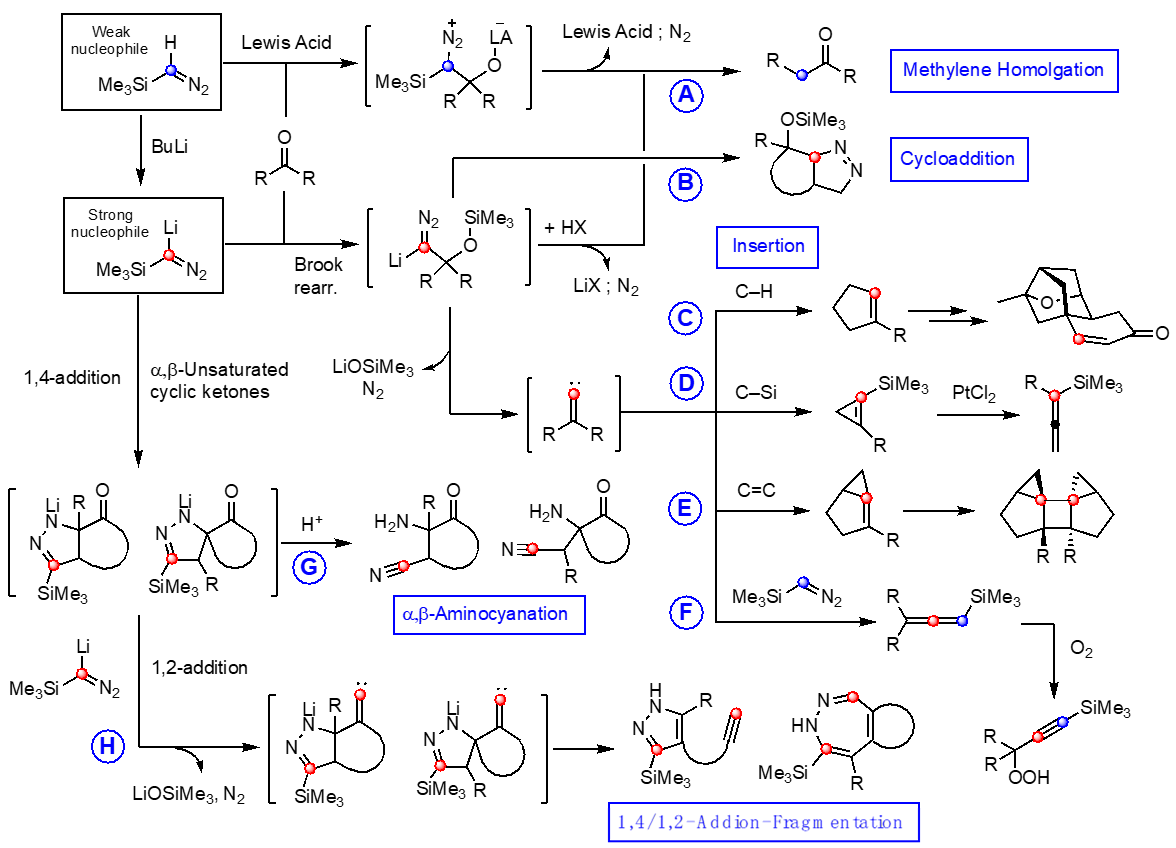
Trimethylsilyldiazomethane (TMSCHN2) has been engaged in various synthetic transformations and we have been exploring the novel reactivity of TMSCHN2 especially in carbon-carbon bond forming reactions, which are graphically outlined. As opposed to a common Lewis acid-catalyzed one-carbon homologation withTMSCHN2, the corresponding lithiated reagent TMSC(Li)N2 is a much stronger nucleophile, thus it reacts readily with a broader range carbonyl compounds to generate one-carbon homologation product with high selectivity (Path A). On the other hand, the same intermediate can undergo an intramolecular cyclization with tethered alkene to generate Δ1-pyrazolines (Path B). By controlling the reaction temperature, the adducts between carbonyl compounds and TMSC(Li)N2 undergo elimination of LiOSiMe3 followed by N2 to generate alkylidene carbenes. This highly reactive unsaturated carbene species can participate in an insertion reaction with a C–H bond to generate cyclopentene derivatives (Path C). The insertion of alkylidene carbene onto a bridgehead C–H was also possible, which was employed as a key strategy in the novel construction of platensimycin core. α-Silyl ketones react with TMSC(Li)N2 to generate an alkylidene carbene intermediate, which readily undergo C–Si bond insertion to generate silyl cyclopropenes, treatment of which with PtCl2 efficiently rearrange the cyclopropene moiety to generate silyl allenes (Path D). Alkylidene carbenes generated from γ,δ-unsaturated ketones undergo addition onto the π-bond to generate highly strained bicyclo[3.1.0]hex-1-ene systems, which readily undergo dimerization (Path E). It was discovered that alkylidene carbenes generated from various acyclic and cyclic ketones readily react withTMSCHN2, providing silyl allenes, which are prone to react with molecular oxygento generate the corresponding propargylic peroxides (Path F). α,β-Unsaturated cyclic ketones react with TMSC(Li)N2 in a 1,4-addition mode followed by cyclization to generate pyrazolines, which lead to non-reductive N–N bond cleavage to generate α,β-aminocyanation products upon treating with TsOH (Path G). These α,β-unsaturated cyclic ketones also react sequentially with 2 equivalents of TMSC(Li)N2. With appropriate structural elements, alkylidene carbenes generated from lithium pyrazolinate intermediate can undergo fragmentation to form pyrazoles containing a tethered alkyne moiety, or alternatively undergo N–L bond insertion followed by cycloreversion of the azete intermediate to provide 1,2-diazepines (Path H). [Top]
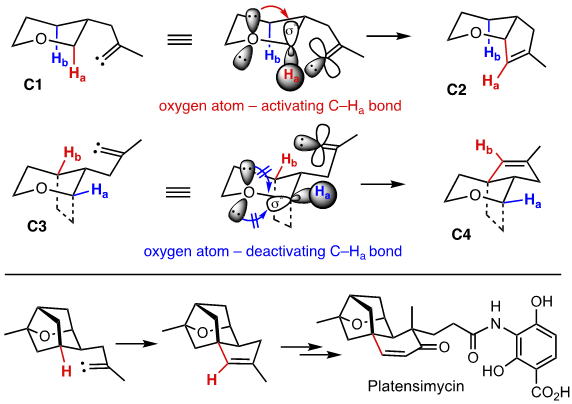
Alkylidene carbene insertion for the synthesis of platensimycin: The fate of alkylidene carbenes depend on the interplay of multiple parameters. One of the ways to steer the alkylidene carbene insertion toward a specific C–H bond is to install an activating group such as an ether functionality to the carbon bearing the C–H bond undergoing the insertion. In general, the activating role of the oxygen can be rationalized by the participation of its lone-pair electrons. However, the extent of relative contribution by oxygen substituents in different environments and the exact mechanism by which these oxygen substituents activate a particular C–H bond toward the insertion are not known. In this regard, we hypothesized that the effect of the oxygen substituent should be manifested by the n(O) to σ*(C–H) electron delocalization. If this is indeed the case, insertion reactions with conformationally constrained systems would unambiguously reveal the stereoelectronic effect of the oxygen, thereby the regio- and stereoselectivity of these reactions will become predictable. This hypothesis was tested by employing probes C1 and C3. As shown, the lone-pair electrons in C1 would activate the axial C–Ha bond, leading to insertion product C2. On the other hand, in C3, due to the poor alignment of the orbitals, the expected n(O) to σ*(C–H) delocalization will be minimal if not zero, thus the oxygen functionality is an electron-withdrawing group because of its strong inductive effect. In this situation, the Ha will become deactivated to the insertion, which would direct the insertion to Hb to generate C4. Indeed, C1 gave exclusively C2, whereas C3 afforded product C4. This strong stereoelectronic effect-based selectivity control of C–H insertion guided us to design a synthesis strategy for platensimycin. [Top]
Based on this plan, we prepared bridged oxabicycle D1 starting from reducing (S)-carvone (LAH, –78 °C) and bromoetherification (NBS, –78 °C). The diastereoselectivity for these two steps are 15:1 and 12:1 respectively. The stereochemistry at the newly formed quaternary center was confirmed by an X-ray crystallographic analysis of D2, which was obtained via allylic oxidation of the methyl group (SeO2 followed by PCC). Cyclization of bromoaldehyde D2 was accrued out by Bu3SnH/AIBN at 65 °C (81%, 4.5:1). One-carbon homologation of D3 via the Wittig reaction and hydrolysis with NBS/zinc powder provided the homologated aldehyde D4. Addition of methyl Grignard reagent followed by oxidation afforded ketone D5. Treatment of ketone D5 with TMSC(Li)N2 generated from BuLi andTMSCHN2 afforded C–H insertion product D6. (KOH in Dihydroxylation of D6, oxidative cleavage of the resulting diol (NaIO4), and intramolecular aldol/dehydration MeOH) provided enone D7 in excellent yield. Following the protocol reported by Nicolaou, the tricyclic core D7 was elaborated to platensimycin. This C–H insertion strategy is currently exploited for the synthesis of other terpene natural products including lungshengenin D, which contain the identical caged structure D7. [Top] 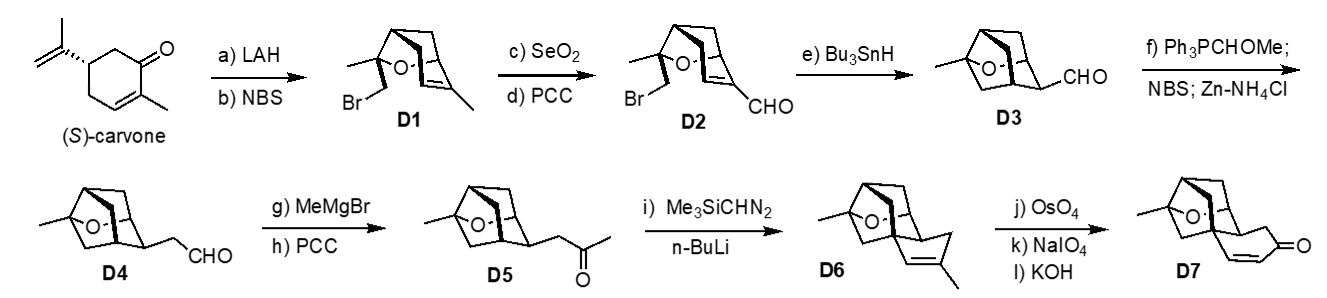
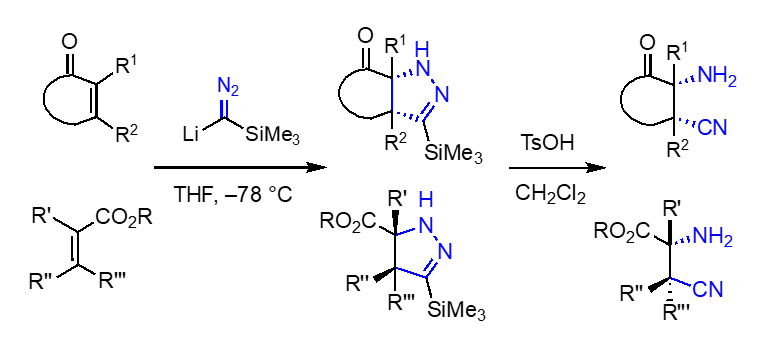
Aminocyanation of α,β-unsaturated esters for the synthesis of amathaspiramides and massadine: α-Amino ketones constitute an important class of biologically active natural products and pharmaceuticals. Conceptually, one of the most straightforward synthetic approaches to the α-amino ketone is α-amination, wherein an enol or enolate of ketones reacts with an electrophilic nitrogen-containing species. Various α-amination methods have been developed, yet most of these methods share a typical shortcoming, which is the tedious manipulation of removing the extra functionality on the nitrogen after C–N bond formation. In this regard, we demonstrated that the addition of TMSC(Li)N2 with α,β-unsaturated ketones to form Δ2-pyrazolines followed by facile proteolytic N–N bond cleavage as an effective method to introduce a free amino group at the α-position of cyclic ketones. This formal aminocyanation reaction of ketones could be extended to α,β-unsaturated esters, which enables the preparation of various structurally novel α-amono esters that contain a cyano group at the β-position. This simple protocol has a significant merit compared to the traditional amination, especially in terms of cleaving a relatively strong N–N bond under non-reductive conditions [Top]
Relying on this aminocyanation, novel strategies for the synthesis of the amathaspiramides and massadine were developed. These synthetic approaches embrace the de novo construction of the quaternary carbon center containing a C–N bond by 1,4-addition of TMSC(Li)N2 with α,β-unsaturated esters. The synthesis of amathaspiramide C and the pivotal intermediate that lead to all members of the amathaspiramide family as well as the core of massadine have been achieved relying on the aminocyanation. Currently, our effort is directed to the completion of massadine total synthesis. [Top] 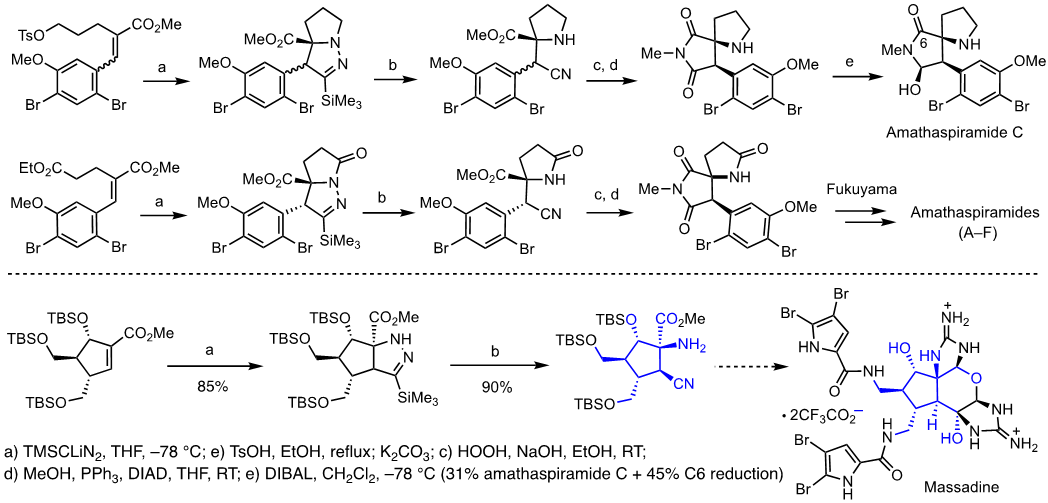
|