Theoretical IR and VCD Studies, primarily applied to Peptide Models

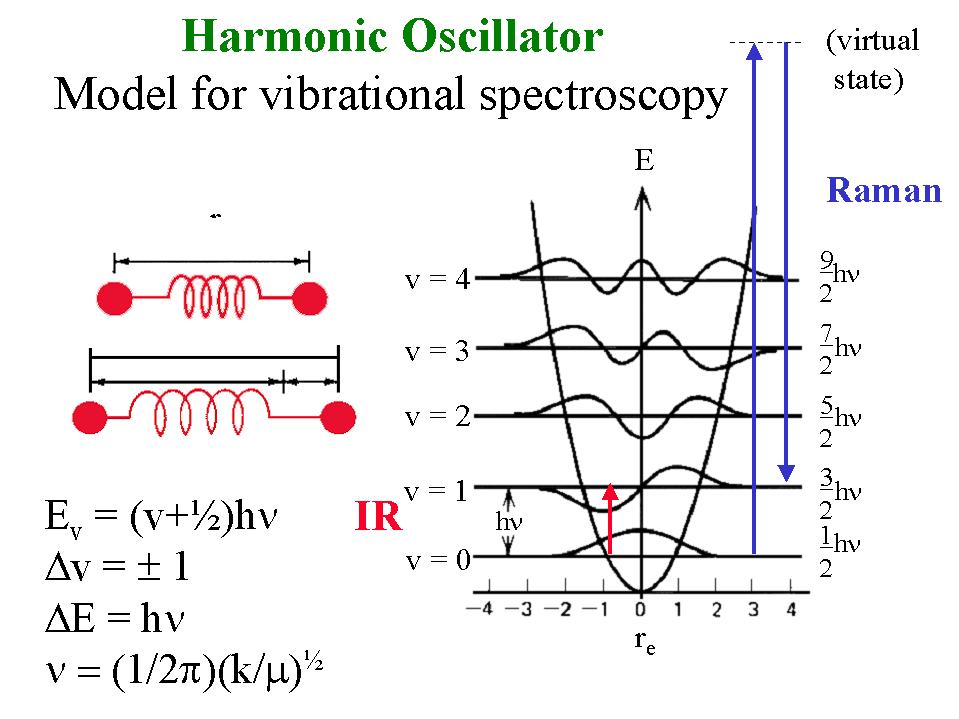
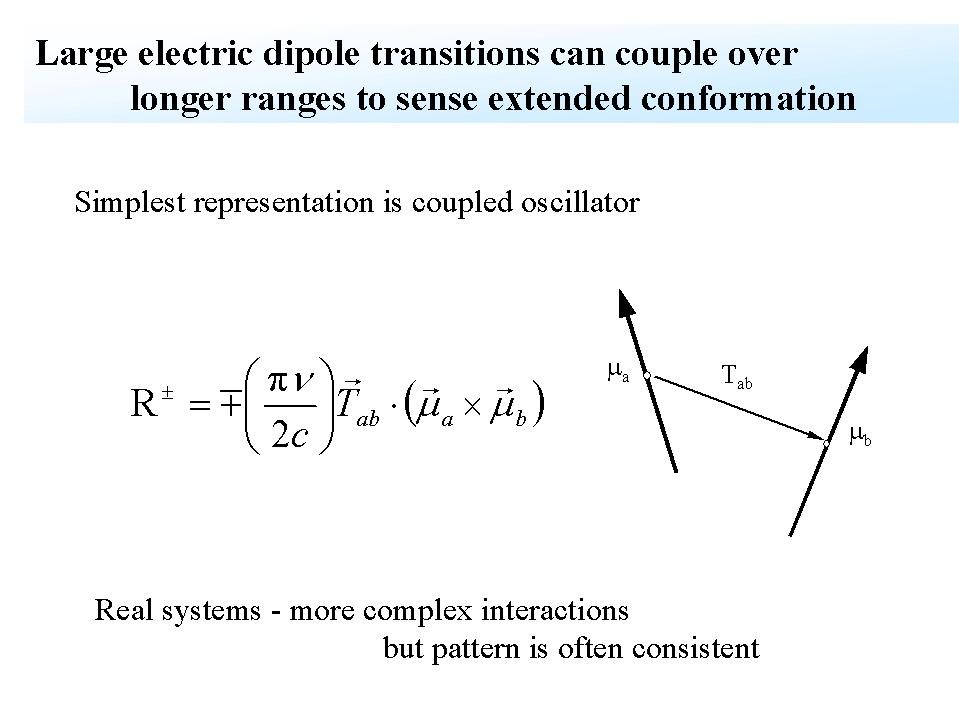

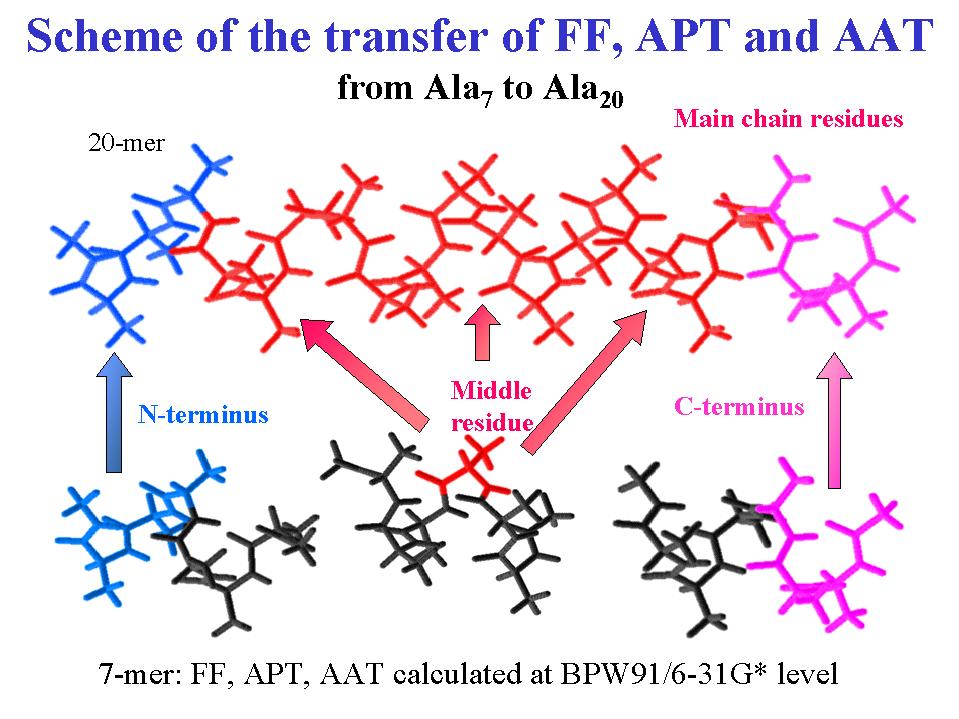
Theoretical basis for VCD, adapted from:
"Protein and Peptide Secondary Structure and Conformational Determination with Vibrational Circular Dichroism"
Timothy A. Keiderling, Current Opinions in Chemical Biology (Ed. Julie Leary and Mark Arnold) 6, 682-688 (2002)
Theoretical Simulations
The major recent development for peptide VCD is the ability to simulate spectra for various conformations based on ab initio quantum mechanical force fields (FF) and intensity parameters, termed atomic polar and axial tensors developed with the magnetic field perturbation (MFP) theory of Stephens (1,2). While such fundamental studies ordinarily do not impact biology, simpler theoretical interpretive models of peptide VCD are unreliable. Since only in ideal cases can well-defined conformations be confidently assigned to small peptides, a reliable theoretical method can be used to explore conformational sensitivity since all conformations are accessible in the computer. Initial studies in this field focused on Gly-based peptides containing two coupled amide functions but constrained to conventional secondary structure geometries (3). Subsequently larger and even solvated small peptides have been modeled (4,5). While peptides of biological interest and proteins are typically too large for such ab initio calculations with current computer capabilities, Bour has developed a method for transfer of FF and intensity parameters from a smaller peptide to a larger one that shares its conformation (5,6). IR and VCD spectra for systems containing as many as 65 residues have been stimulated in this manner . An alternative method using neural networks to correlate computed VCD with conformation for various structures has recently been reported (7). The important issue of solvent effects is also beginning to be addressed theoretically for peptide spectra (8,9).
Initial studies focused on helices and used tripeptides for transfer to larger oligomers , but better results were obtained by using oligomers containing 7 or 5 amides to model a and 310-helices, respectively (5). While the spectra are very similar to those simulated with tripeptides, with this larger sized "small" peptide the central residue is fully hydrogen bonded and thus better coupled to other residues. Accurate simulations of a -helical and left-handed 31-helical (PLP II-like) amide I and II absorption and VCD were obtained. The a -helical low-energy weak negative VCD amide I component depended on length, but the 31-helical result was not, probably due to the lack of internal hydrogen bonds. The qualitative difference between a - and 310-helical VCD and even the dependence on composition (Aib vs. Ala) could be simulated (10). That such subtle effects, due to the electronic structure effects of -methylation, can be modeled points to the simulation's accuracy.
Simulations of thermal denaturation of a -helical peptides with various isotope substitutions showed the C-terminal region to be unwound (not a -helical) and confirmed the utility of modeling the random-coil, thermally denatured state as an extended helix (11). At minimum, two sequential isotope substitutions are needed to develop a helical VCD and separation of the labels by an unlabeled residue weakens the 13C VCD and distorts its sign pattern.
Parallel and anti-parallel b -sheet conformations were the subject of another theoretical study which indicated similar weak predominately negative VCD when both were twisted (12). On the other hand, extended, flat (long multi-stranded) antiparallel b -sheet structures have unique IR and VCD patterns, with an intense low frequency component (IR), such as that often found for model polypeptide b-sheets such as high pH PLL and for aggregated peptides and proteins. This unique structure also has a position sensitive, intense 13C pattern when isotope substituted on alternate residues which acts as a site-specific amplification effect suggesting potential for detecting local, initial anti-parallel sheet formation (13). Extending these methods to hairpins required a modified transfer method to encompass multiple small molecules, which yields spectra in qualitative agreement for the amide I with VCD for model hairpins based on D-Pro-Gly type Ií and IIí turns (14).
Reference
1 Stephens PJ, Devlin FJ,Amouche A: Vibrations of chiral molecules 2: Determination of the structures of chiral molecules using vibrational circular dichroism. In Chirality: Physical Chemistry. ACS Symposium Series, vol 810, edited by J. M. Hicks. New York: Oxford University Press; 2002, 18-33. [The authors present a compact summary of their DFT/GIAO method of computing VCD spectra with the MFP theory and apply it to the simulations of spectra for molecules ranging from methyl oxirane (a small, rigid test case) to large species like Trogler's base and flexible cases containing rotational isomers.]
2. Stephens PJ, Devlin FJ, : Determination of the structure of chiral molecules using ab initio vibrational circular dichroism spectroscopy, Chirality. 2000, 12:172-179.
3. Bour P,Keiderling TA: Ab initio simulation of the vibrational circular dichroism of coupled peptides. J. Am. Chem. Soc. 1993, 115:9602-9607.
4. Bour P, Kubelka J,Keiderling TA: Simulations of oligopeptide vibrational circular dichroism. Effects of isotopic labeling. Biopolymers 2000, 53:380-395.
5. Kubelka J, Silva RAGD, Bour P, Decatur SM,Keiderling TA: Chirality in Peptide Vibrations. Ab Initio Computational Studies of Length, Solvation, Hydrogen Bond, Dipole Coupling and Isotope Effects on Vibrational CD. In Chirality: Physical Chemistry. ACS Symposium Series, vol , edn . edited by J. M. Hicks. New York: Oxford University Press; 2002:50-64.
6. Bour P, Sopkova J, Bednarova L, Malon P,Keiderling TA: Transfer of molecular property tensors in Cartesian coordinates: A new algorithm for simulation of vibrational spectra. J. Comput.Chem. 1997, 18:646-659.
7. Bohr HG, Frimand K, Jalkanen KJ, Nieminen RM,Suhai S: Neural-network Analysis of the Vibrational Spectra of N-acetyl L-alanyl N'-methyl Amide Comformational States. Physical Review E. 2001, 64:1905-.
8. Kubelka J,Keiderling TA: Ab initio calculation of amide carbonyl stretch vibrational frequencies in solution with modified basis sets. 1. N-methyl acetamide. Journal of Physical Chemistry 2001, 105:10922-10928
9. Han WG, Elstner M, Jalkanen KJ, Frauenheim T,Suhai S: Hybrid SCC-DFTB/molecular Mechanical Studies of H-bonded Systems and of N-acetyl-(L-Ala) (n) N'-methylamide Helices in Water Solution Source. International Journal of Quantum Chemistry 2000, 78:459-479
10. Kubelka J, Silva RAGD,Keiderling TA: Spectroscopic Discrimination Between Peptide 310- and a-helices. Study of the Impact of a-Methyl Substitution. J. Am. Chem. Soc. In Press, 2002, 000-000 [VCD and IR of (Ala)2n, (Aib-Ala)n, and (Aib)2n oligomers are calculated for both a- and 310-helical conformations and compared to data for Ala-rich, Aib-rich and Aib-Ala mixed oligomers. The theoretical results qualitatively predict the small intensity shifts with increase in Ala content that occur in the 310 helix form and confirm that the major intensity differences in VCD between a- and 310-helices is not due to aMe]
11. Silva RAGD, Kubelka J, Decatur SM, Bour P,Keiderling TA: Site-Specific Conformational Determination in Thermal Unfolding Studies of Helical Peptides using Vibrational Circular Dichroism with Isotopic Substitution. Proc. Natl. Acad. Sci. U. S. A. 2000, 97:8318-8323
12. Kubelka J,Keiderling TA: Differentiation of beta-sheet-forming structures: Ab initio-based simulations of IR absorption and vibrational CD for model peptide and protein beta-sheets. J. Am. Chem. Soc. 2001, 123:12048-12058.
13. Kubelka J,Keiderling TA: The anomalous infrared amide I intensity distribution in C-13 isotopically labeled peptide beta-sheets comes from extended, multiple-stranded structures. An ab initio study. J. Am. Chem. Soc. 2001, 123:6142-6150.
14. Hilario J, Kubelka J, Syud FA, Gellman SH,Keiderling TA: Spectroscopic Characterization of Selected b-Sheet Hairpin Models. Biospectroscopy 2002,